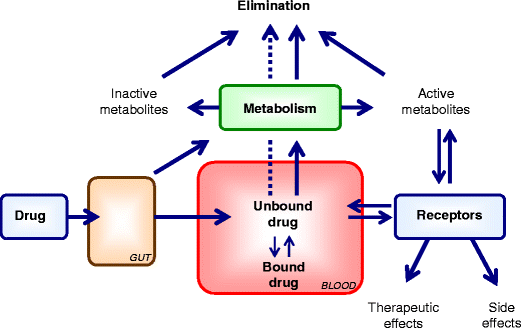
Protein Binding
Many drugs interact with plasma or tissue proteins or other macromolecules, such as melamine or DNA, to form a drug- macromolecule complex. The formation of a drug protein complex is often named as drug-protein binding. Drug protein binding may be a reversible or irreversible process. The formation of drug protein complex is named Drug Protein Binding.
Drug- Protein binding may be a reversible or an irreversible process.
- Irreversible protein binding: due to chemical activation of the drug which then attaches strongly to the protein by covalent bond. It may result in certain type of drug toxicity that may occur over a long time such as carcinogenesis, or within a short time as the formation of reactive chemical intermediates.
- Reversible protein binding: drug binds with the proteins with weaker chemical bonds like Hydrogen bonds or Vander Waals forces.
Most drugs bind with proteins by reversible process.
Amino acid that composes the protein chain have hydroxyl, carboxyl, or other sites available for reversible drug interaction. The protein bound drugs is a large complex that cannot easily transverse the cell or capillary membrane and therefore has a restricted distribution. Protein bound drug is pharmacologically inactive, in contrast free or unbound drug crosses the cell membrane and is therapeutically active. Purified protein such as albumin is used to evaluate drug-protein binding. Methods such as equilibrium dialysis and ultrafiltration make use of semipermeable membrane that separate protein and protein bound drug from free or unbound drug. By these in vitro methods, the conc. of bound drug, free drug and total protein may be determined.
Different in vitro methods have advantages and disadvantages in terms of
- Cost
- Ease of measurement
- Time
- Instrumentation
- Other considerations
Factors affecting protein binding:
The Drug
- Physicochemical properties and concentration of the drug in body.
- Increase in lipophilicity, increase extend of binding e.g., cloxacillin 95% bound, ampicillin 20% bound.
- Acidic/anionic drugs bind to albumin.
- Basic/cationic drugs bind to globulins.
- Neutral/unionized drugs bind to lipoproteins.
- At low concentration, most drugs bound to the proteins, but at higher concentration more free drugs may be present owing to saturation of binding sites on protein.
The Protein
- Quality or physicochemical nature of the protein synthesized.
- Concentration of the protein available for drug protein binding.
- Number of binding sites on the proteins.
- Concentration is controlled by variables such as synthesis, catabolism, distribution between intra and extravascular space and number of diseases
Competition between the drug and body constituents
- Interaction of drugs with free fatty acids.
- Free fatty acid levels are increased during fasting, diabetes, myocardial infarction etc.
- For example: Interaction of sodium salicylate and bilirubin in neonates.
Drug Interactions
- Competition of drug by other substances at a protein binding site e.g., phenylbutazone and warfarin when given simultaneously, warfarin is displaced by phenylbutazone.
- Alteration of the protein structure by the drug metabolite that modifies binding capacity. e.g., aspirin acetylation of albumin, modify the binding capacity of NSAIDs.
The pathophysiologic conditions of the patient
- Liver disease: decrease in albumin conc. due to decreased synthesis.
- Nephrotic syndrome: due to accumulated metabolites urea and uric acid, the protein binding may be altered
- Severe burns: increased distribution of albumin in ECF.
- Genetic disorders: the quality of the protein may be altered due to genetic disorders which may result in decreased affinity for drug
Speed of Injection
- Rapid IV injection may increase the free drug concentration of some highly protein bound drugs and therefore increase in intensity of its action.
- It Can be attributed to initial saturation of the protein binding sites.
- For example: Diazoxide a hypotensive drug.
- Injection time: 10 sec dramatic increased hypotensive effect.
- Injection time: 100 sec smaller hypotensive effect.